著名鋼琴家貝多芬在32歲時聽力完全喪失,其傷害不言而喻�����,他在留給其兄弟的遺囑中寫道�����,“正如落葉凋零,我的生命也已變得空洞����。”
盡管貝多芬聽力喪失的原因未知�����,但很多案例顯示聽力障礙實(shí)際都和遺傳DNA突變有關(guān)����,而目前已知的和聽力障礙相關(guān)的基因有近100個。但迄今為止��,幾乎沒有任何治療方法可以減緩或者逆轉(zhuǎn)聽力障礙�。貝多芬逝世兩個世紀(jì)后的今天,阻止遺傳因素導(dǎo)致聽力障礙的技術(shù)離實(shí)現(xiàn)臨床已越來越近����。
哈佛大學(xué)醫(yī)學(xué)院耳鼻喉科副教授Zheng-Yi Chen和哈佛大學(xué)化學(xué)與化學(xué)生物學(xué)教授��、美國博德研究所核心成員David R. Liu共同主導(dǎo)的一項(xiàng)研究在線發(fā)表在在國際著名期刊《自然》(Nature)�����,該團(tuán)隊(duì)用基因編輯技術(shù)治療TMC1突變“貝多芬小鼠”的遺傳性聽力障礙,并取得良好效果�。
內(nèi)耳是聲音感知的最重要部位,其中內(nèi)耳毛細(xì)胞又是人類聽到聲音的關(guān)鍵���。其中��,TMC1是內(nèi)耳毛細(xì)胞中機(jī)械力傳導(dǎo)的重要組成部分��,TMC1蛋白在內(nèi)耳毛細(xì)胞的纖毛上形成通道���,當(dāng)聲波引起纖毛運(yùn)動時這種通道就會打開,隨后鈣離子進(jìn)入細(xì)胞生成電信號���,電信號傳遞到大腦后形成聽覺��。
TMC1發(fā)生了一種顯性負(fù)性錯義突變后����,會導(dǎo)致內(nèi)耳毛細(xì)胞單通道電流水平和鈣滲透率降低�,進(jìn)一步導(dǎo)致感音神經(jīng)性語后聾(語言形成后出現(xiàn)聽力障礙)。通常�����,TMC1顯性突變患者在10-15歲時會開始逐漸變聾。
TMC1顯性突變即意味著���,一對等位基因中只有一個發(fā)生突變就會導(dǎo)致功能喪失��,從而致聾���。這也就使得修復(fù)顯性突變成了一項(xiàng)精細(xì)的任務(wù):必須使突變基因失活,同時還要保留野生型基因����。這一對基因的差別只在于其中一個核苷酸的不同。突變的TMC1在原本野生型一個胸腺嘧啶核苷酸T的位置變成了腺嘌呤核苷酸A����。
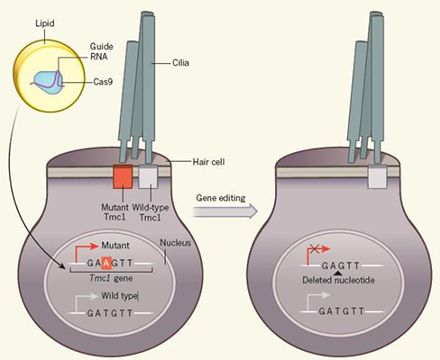
基因編輯過程示意圖
Chen和Liu團(tuán)隊(duì)啟用的是熱門的CRISPR–Cas基因編輯技術(shù)。研究團(tuán)隊(duì)將含有Cas9和RNA的脂滴注入新生TMC1貝多芬小鼠的內(nèi)耳��,這種脂滴能和細(xì)胞隨后融合�。基因編輯后突變基因中腺嘌呤核苷酸A被敲除�����,從而使該基因失活�。但同時保留了野生型基因,保證正常聽力功能實(shí)現(xiàn)�����。
研究團(tuán)隊(duì)評估得出�,基因編輯過的小鼠中內(nèi)耳毛細(xì)胞的存活率更高、聽腦干反應(yīng)閾值更低����。此外,聽覺反射也出現(xiàn)改善��,基因編輯過的成年貝多芬小鼠在聽到突發(fā)噪音時會表現(xiàn)出驚嚇�,然而未經(jīng)基因編輯的小鼠對此毫無反應(yīng)。
論文最后指出���,這一治療途徑在治療和內(nèi)耳毛細(xì)胞功能障礙有關(guān)的常染色體顯性遺傳聽力障礙方面具備潛力�����,也為反義寡核苷酸療法和RNA干擾療法提供了補(bǔ)充策略��。
更多詳情請登錄聽覺有道助聽器cxji.cn